反转录PCR(以mRNA为模板合成的PCR)

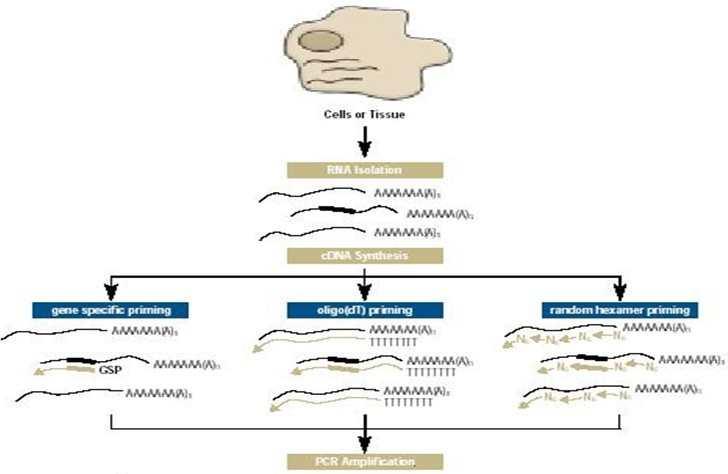
反转录PCR (reverse transcription PCR,RT-PCR),是指以mRNA为模板 在反转录酶作用下合成的cDNA 第一链的PCR。该反应分为两步,第一步在单引物的介导和反转录酶的催化下合成RNA 的互补链cDNA; 第二步加热后cDNA 与RNA 链解离,然后与另一引物退火,并由DNA 聚合酶催化引物延伸生成双链DNA,最后同常规PCR一样,扩增靶DNA。合成cDNA 第一链所用的反转录酶是一类依赖于RNA 的DNA 聚合酶。目前常用的反转录酶主要有两种:一种是来源于禽成髓细胞性白血病病毒(avian myeloblastosis virus,AMV)的AMV 反转录酶,另一种是来源于莫洛尼鼠白血病病毒(moloney murine leukemia virus,MoMLV) 的MOMLV 反转录酶。 反转录PCR(Reverse Transcription,RT-PCR)又称为逆转录PCR。其原理是:提取组织或细胞中的总RNA,以其中的mRNA作为模板,采用Oligo(dT)或随机引物利用逆转录酶反转录成cDNA。再以cDNA为模板进行PCR扩增,而获得目的基因或检测基因表达。
反转录PCR
Reverse Transcription PCR,RT-PCR
逆转录PCR
提取组织或细胞中的总RNA
聚合酶链式反应广泛应用的变形
概述

RT-PCR反应原理
RT-PCR使RNA检测的灵敏性提高了几个数量级,使一些极为微量RNA样品分析成为可能。该技术主要用于:分析基因的转录产物、获取目的基因、合成cDNA探针、构建RNA高效转录系统。 RT- PCR即逆转录PCR,是将RNA的逆转录(RT)和cDNA的聚合酶链式扩增反应(PCR)相结合的技术。RT-PCR技术灵敏而且用途广泛,可用于检测细胞/组织中基因表达水平,细胞中RNA病毒的含量和直接克隆特定基因的cDNA序列等。选择
1. Money 鼠白血病病毒(MMLV)反转录酶:有强的聚合酶活性,RNA酶H活性相对较弱。最适作用温度为37℃。
2. 禽成髓细胞瘤病毒(AMV)反转录酶:有强的聚合酶活性和RNA酶H活性。最适作用温度为42℃。
3.Thermus thermophilus、Thermus flavus等嗜热微生物的热稳定性反转录酶:在Mn2 存在下,允许高温反转录RNA,以消除RNA模板的二级结构。
4.MMLV反转录酶的RNase H-突变体:商品名为SuperScript 和SuperScriptⅡ。此种酶较其它酶能多将更大部分的RNA转换成cDNA,这一特性允许从含二级结构的、低温反转录很困难的mRNA模板合成较长cDNA。
合成
1. 随机六聚体引物:当特定mRNA由于含有使反转录酶终止的序列而难于拷贝其全长序列时,可采用随机六聚体引物这一不特异的引物来拷贝全长mRNA。用此种方法时,体系中所有RNA分子全部充当了cDNA第一链模板,PCR引物在扩增过程中赋予所需要的特异性。通常用此引物合成的cDNA中96%来源于rRNA。
2. Oligo(dT):是一种对mRNA特异的方法。因绝大多数真核细胞mRNA具有3’端Poly(A )尾,此引物与其配对,仅mRNA可被转录。由于Poly(A )RNA仅占总RNA的1-4%,故此种引物合成的cDNA比随机六聚体作为引物和得到的cDNA在数量和复杂性方面均要小。
3. 特异性引物:最特异的引发方法是用含目标RNA的互补序列的寡核苷酸作为引物,若PCR反应用二种特异性引物,第一条链的合成可由与mRNA 3’端最靠近的配对引物起始。用此类引物仅产生所需要的cDNA,导致更为特异的PCR扩增。
操作
1. 总RNA的提取。
2. cDNA第一链的合成(Reverse Transcription):如今试剂公司有多种cDNA第一链试剂盒出售,其原理基本相同,但操作步骤不一。现以GIBICOL公司提供的SuperScriptTM Preamplification System for First Strand cDNA Synthesis 试剂盒为例。
(1)在0.5ml微量离心管中,加入总RNA 1-5μg,补充适量的DEPC H2O使总体积达11μl。在管中加10μM Oligo(dT)12-18 1μl,轻轻混匀、离心。
(2)70℃加热10min,立即将微量离心管插入冰浴中至少1min。
然后加入下列试剂的混合物:
10×PCR buffer 2μl
25mM MgCl2 2μl
10mM dNTPmix 1μl
0.1M DTT 2μl
轻轻混匀,离心。42℃孵育2-5min。
(3)加入SuperscriptⅡ1μl ,在42℃水浴中孵育50min。
(4)于70℃加热15min以终止反应。
(5)将管插入冰中,加入RNase H 1μl ,37℃孵育20min,降解残留的RNA。-20℃保存备用。
3.PCR:
(1)取0.5ml PCR管,依次加入下列试剂:
第一链cDNA 2μl
上游引物(10pM) 2μl
下游引物(10pM) 2μl
dNTP(2mM) 4μl
10×PCR buffer 5μl
Taq 酶(2u/μl) 1μl
(2)加入适量的ddH2O,使总体积达50μl。轻轻混匀,离心。
(3)设定PCR程序。在适当的温度参数下扩增28-32个循环。为了保证实验结果的可靠与准确,可在PCR扩增目的基因时,加入一对内参(如G3PD)的特异性引物,同时扩增内参DNA,作为对照。
(4)电泳鉴定:行琼脂糖凝胶电泳,紫外灯下观察结果。
(5)密度扫描、结果分析:采用凝胶图像分析系统,对电泳条带进行密度扫描。
注意
1. 在实验过程中要防止RNA的降解,保持RNA的完整性。在总RNA的提取过程中,注意避免mRNA的断裂。
2. 为了防止非特异性扩增,必须设阴性对照。
3. 内参的设定:主要为了用于靶RNA的定量。常用的内参有G3PD(甘油醛-3-磷酸脱氢酶)、β-Actin(β-肌动蛋白)等。其目的在于避免RNA定量误差、加样误差以及各PCR反应体系中扩增效率不均一各孔间的温度差等所造成的误差。
4. PCR不能进入平台期,出现平台效应与所扩增的目的基因的长度、序列、二级结构以及目标DNA起始的数量有关。故对于每一个目标序列出现平台效应的循环数,均应通过单独实验来确定。
5. 防止DNA的污染:
(1)采用DNA酶处理RNA样品。
(2)在可能的情况下,将PCR引物置于基因的不同外显子,以消除基因和mRNA的共线性。