全基因组测序(对未知基因组物种进行个体测序)

VLoG
次浏览
更新时间:2023-05-19
全基因组测序
对未知基因组物种进行个体测序
全基因组测序是对未知基因组序列的物种进行个体的基因组测序。 1986年, Renato Dulbecco是最早提出人类基因组测序的科学家之一。他认为如果能够知道所有人类基因的序列,对癌症的研究将会很有帮助。美国能源部(DOE)与美国国家卫生研究院(NIH),分别在1986年与1987年加入人类基因组计划。除了美国之外,日本在1981年就已经开始研究相关问题,但是并没有美国那样积极。到了1988年,詹姆士·华生(DNA双螺旋结构发现者之一)成为NIH的基因组部门主管。1990年开始国际合作。1996年,多个国家召开百慕达会议,以2005年完成测序为目标,分配了各国负责的工作,并且宣布研究结果将会及时公布,并完全免费。
基本信息
| 中文名 | 全基因组测序 |
| 测序目标 | 2005年 |
| 准备时间 | 19世纪80-90年代 |
| 国际合作 | 1990年 |
研究结果
①NCI-H209细胞系基因组中,共检测到22,910个碱基替换、65个插入缺失(Indels)、58个结构变异;在基因组的编码区,除了发现RB1 和TP53基因发生点突变和MLL2基因由于发生了G>T的颠换,从而产生了pre-stop codon外,有94个点突变直接改变了氨基酸序列,有36个属同义突变。
②特定的碱基及其周围序列易被烟气中的多环芳烃和丙烯醛诱变。在NCI-H209细胞系基因组中,G>T/C>A是最为普遍的颠换现象,发生频率为34%;其次是G>A/C>T(21%)和A>G/T>C(19%);CpG岛外的CpG二核苷酸多发生G>T颠换,而CpG岛内的CpG二核苷酸多发生G>C颠换,说明烟气中的致癌物偏好引起甲基化的CpG二核苷酸发生颠换。
③检测到转录偶联修复(Transcription-coupled repair)和表达相关的修复(Expression-linked repair)在起作用。转录偶联修复作用机制:鸟嘌呤和腺嘌呤上大的加合物是吸烟过程中所释放的致癌化学物质引起DNA损伤的主要形式,这些大的加合物阻止了转录链上RNA聚合酶的转录过程,而转录受阻的RNA聚合酶招募核苷酸剪切修复相关因子对受损的核苷酸进行修复以避免突变发生。在TP53基因突变的肺癌细胞中,G>T颠换常出现在非转录链,表明在转录链上相同的损伤已被识别和修复。在本研究中,转录链上G和A碱基替换频率比非转录链上少,由此看来嘌呤是烟气致癌物质主要诱变靶标。另外,在NCI-H209细胞系中,转录链和非转录链上发生不同类型的突变(G>T、A>G、A>T)两条链基因表达水平也有差异,这就意味着转录偶联修复机制识别、修复不同加合物损伤的能力不同。
表达相关的修复(Expression-linked repair)作用机制:这是一种新的、更为普遍的修复机制,即,高表达的基因中,转录链及非转录链的突变频率都较低。在NCI-H209细胞系中,转录链和非转录链上发生G>A的突变,两条链上基因表达水平都很高,这就说明表达相关的修复作用比转录偶联修复作用更为重要。
简介
技术路线
原理
双末端(Paired-End)测序原理
测序深度(SequencingDepth):测序得到的碱基总量(bp)与基因组大小(Genome)的比值,它是评价测序量的指标之一。测序深度与基因组覆盖度之间是一个正相关的关系,测序带来的错误率或假阳性结果会随着测序深度的提升而下降。重测序的个体,如果采用的是双末端或Mate-Pair方案,当测序深度在10~15X以上时,基因组覆盖度和测序错误率控制均得以保证。
测序深度对基因组覆盖度和测序错误率的影响
全基因组重测序的个体,通过序列比对,可以找到大量的单核苷酸多态性(SNP),插入缺失(InDel,Insertion/Deletion)和结构变异(SV,StructureVariation)位点。SBC可以协助客户,通过生物信息手段,分析不同个体基因组间的结构差异,同时完成SNP及基因组结构注释。
测序覆盖度:基因组被测序得到的碱基覆盖的比例;测序覆盖度是反映测序随机性的指标之一;测序序深度与覆盖度之间的关系可以过Lander-Waterman Model(1988)来确定。当深度达到5X时,则可覆盖基因组的约99.4%以上。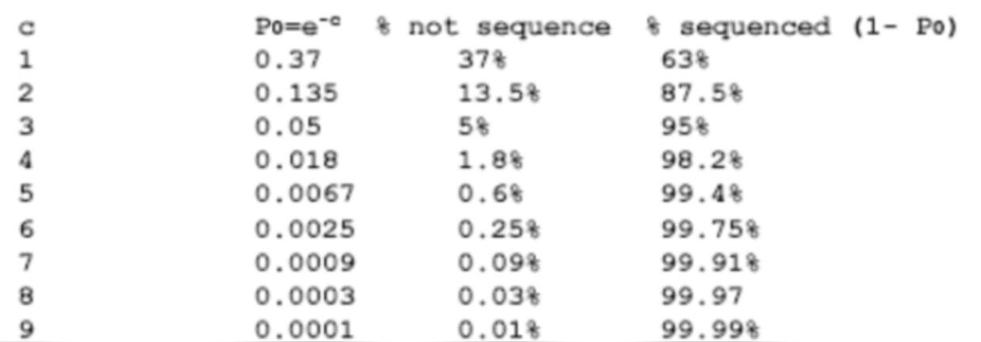

全基因组测序
分析流程
1.数据量产出
总碱基数量、TotalMappingReads、UniquelyMappingReads统计,测序深度分析。
2.一致性序列组装
3.SNP检测及在基因组中的分布
提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。并根据参考基因组信息对检测到的变异进行注释。
4.InDel检测及在基因组的分布
在进行mapping的过程中,进行容gap的比对并检测可信的shortInDel。在检测过程中,gap的长度为1~5个碱基。对于每个InDel的检测,至少需要3个Paired-End序列的支持。
5.StructureVariation检测及在基因组中的分布
SBC能够检测到的结构变异类型主要有:插入、缺失、复制、倒位、易位等。根据测序个体序列与参考基因组序列比对分析结果,检测全基因组水平的结构变异并对检测到的变异进行注释。