药物代谢动力学(研究机体对药物处置变化的学科)

VLoG
次浏览
更新时间:2023-05-22
药物代谢动力学
研究机体对药物处置变化的学科

药物代谢动力学(pharmacokinetics)简称药代动学或药动学,主要研究机体对药物的处置(Dispostion) 的动态变化。包括药物在机体内的吸收、分布、生化转换(或称代谢) 及排泄的过程,特别是血药浓度随时间变化的规律。药物的代谢与人的年龄、性别、个体差异和遗传因素等有关。
基本信息
| 中文名 | 药物代谢动力学 |
| 外文名 | pharmacokinetics |
| 别名 | 药代动学或药动学 |
| 方法 | 数学原理和方法 |
简介
药物代谢动力学
包括药物消除动力学
一级消除动力学:单位时间内消除的药量与血浆药物浓度成正比,又叫恒比消除
零级消除动力学:单位时间内体内药物按照恒定的量消除,又叫恒量消除
药物代谢动力学的重要参数:
1、药物清除半衰期(half life),是血浆药物浓度下降一半所需要的时间。其长短可反映体内药物清除速度。
2、清除率(clearance,CL),是机体清除器官在单位时间内清除药物的血浆容积,即单位时间内有多少毫升的血浆中所含药物被机体清除。
4、生物利用度(bioavailability),即经任何给药途径给予一定剂量的药物后到达全身血液循环内药物的百分比。
基本结构
药物转运
药物经膜孔扩散和脂溶扩散通过生物膜。特点:由高浓度向低浓度扩散,直至膜两侧浓度相等(动态平衡);不需酶,不耗能;无饱和现象,也不受其它转运物质抑制;多属外源性物质的转运方式。被动转运包括膜孔扩散和脂溶扩散。膜孔扩散(滤过):膜孔直径约8Å(埃,1Å=10米),直径<8Å或分子量<100的水溶性小分子物质均易通过膜孔扩散。脂溶扩散:即非离子扩散,细胞膜具有类脂结构,脂溶性药物可溶于类脂质透过细胞膜,药物的脂溶性越大越易扩散。扩散速度取决于膜两侧药物浓度梯度及药物在膜内的溶解度。受药物解离度的影响也很大。药物离解成阴、阳离子后,极性增加,脂溶性下降,难穿透类脂质屏障。
(二)特殊转运:主要包括主动转运,易化扩散和膜泡运输。主动转运又名“上坡”转运:特点:是一种载体转运,靠酶促,耗能;可逆浓度梯度透过细胞膜;两种药物转运机制相同时,可出现竞争性抑制;有饱和现象;多属内源性代谢物质的转运方式。易化扩散:是通过镶嵌在细胞膜上的多肽蛋白质来进行的。药物与膜蛋白外侧亚单位(载体)结合后,引起该蛋白质构型改变,将药物甩向内侧,再由该蛋白质的内侧亚单位通过构型变化,进一步把药物甩入细胞内。与主动转运不同之处是顺浓度梯度,不需酶促,不耗能;所需载体在药物浓度高时可被饱和,转运系统可被某些物质抑制或竞争。细胞表面内陷,优质膜把环境物质包围成小泡,再脱离膜质进入细胞内的过程,为内吞作用;细胞内的细胞质小泡同膜质融合,把其所含物质排除细胞,为外排作用。
(三)载体运转
三、药物的体内过程:即药物被吸收进入机体到最后被机体排出的全部历程,包括吸收、分布、代谢和排泄等过程。其中吸收、分布和排泄属物理变化称为转运。代谢属于化学变化亦称转化。机体对药物作用的过程,表现为体内药物浓度随时间变化的规律。药物动力学是研究药物体内过程规律,特别是研究血药浓度随时间而变化的规律。
(一)吸收:药物从给药部位进入血循环称为吸收。影响吸收的因素主要有: 1、给药途径:吸收速度:吸入>舌下>肌注>皮下>直肠>口服>皮肤。 2、药物性质: (1)脂溶性:脂溶性越大,吸收越快; (2)水溶性:易溶于水的药物易吸收; (3)离解度:不解离部分脂溶性较大,易吸收;而解离部分,由于带有极性,脂溶性低,难以吸收。。口服药物被吸收进入体循环的比率,即给药量与吸收量的比率称为生物利用度(或生物可用度)。
(二)分布:影响药物分布的主要因素有: 1、药物的性质:脂溶性大分布到组织器官的速度快。 2、药物与组织的亲和力:有些药物对某些组织器官有特殊的亲和力。药物对组织器官的亲和力与疗效及不良反应有关。 3、药物与血浆蛋白(主要是白蛋白)结合率:结合率大小与疗效有关。结合后: (1)无活性; (2)不易透过毛细血管壁,影响分布和作用; (3)结合型药物分子量大,不易从肾小球滤过,也不受生物转化的影响;因此在体内的作用时间也延长。 4、血流量大小:脑、心肝、肾等组织器官血管丰富,血流量大,药物浓度较高,有利于发挥作用,也易引起这些组织器官损害。 5、特殊屏障:血脑屏障是血液与脑组织之间的屏障,极性小而脂溶性大的药物较易通过,对极性大而脂溶性小的药物则难以通过。
(三)代谢(生物转化)药物代谢的主要器官是肝脏。也可发生在血浆、肾、肺、肠及胎盘。1、药物代谢(转化)酶:(1)肝微粒体药酶:药物在体内主要靠肝细胞微粒体的药酶。其中最主要的是混合功能氧化酶系,其由三部分组成:血红蛋白类,包括细胞色素P-450及细胞色素b5;黄素蛋白类,包括还原型辅酶Ⅱ-细胞色素C还原酶(或称还原型辅酶Ⅱ-细胞色素P-450还原酶)及还原型辅酶I-细胞色素b5还原酶,是电子传递的载体;脂类,主要是磷脂酰胆碱,功能尚不清楚。此三部分共同构成电子传递体系,使用使药物氧化,三者缺一,药物代谢就不能完成。 (2)细胞浆酶系:包括醇脱氢酶、醛氧化酶、黄嘌呤氧化酶等。一些药物经微粒体药酶氧化生成醇或醛后,再继续由醇脱氢酶和醛氧化酶代谢。 (3)线粒体酶:包括单胺氧化酶、脂环族芳香化酶等。单胺氧化酶能使各种内源性单胺类(多巴胺、肾上腺素、去甲肾上腺素、5-羟色胺等)和外源性的胺类(乳酪或酵母中的酪胺等)氧化脱氨生成醛,再进一步氧化灭活。 (4)血浆酶系:包括单胺氧化酶、儿茶酚胺氧位甲基转移酶、酰胺酶及假胆碱酯酶等。前二者可氧化血浆中内源性或外源性单胺类物质。 (5)肠道菌丛酶系:能将某些营养物质变为胺类、羧酸或烃类等有毒物质,肠道菌大量繁殖,产胺过多,可能诱发严重肝功不良者的昏迷,故临床上口服新霉素的目的是杀灭肠道菌丛减少胺类生成,从而减轻肝昏迷。2、代谢(转化)类型:可分两类。第一类包括氧化、还原及水解过程;第二类为结合过程,第一类转化产物再经与体内某些代谢物结合,产物一般水溶性加大,利于排泄。 (1)第一阶段反应(第一类型):氧化、还原及水解等。氧化,如醇氧化、醛氧化、单胺氧化、氧化脱氢及N-氧化等;还原,如硝基还原成氨基(-NH2)。 (2)第二阶段反应(第二类型):即结合反应,使药失效,随尿排出。含羟基、羧基、胺基的化合物与葡萄糖醛酸结合成酯、醚、酰胺化合物;硫酸可与酚类药物及酚性类固醇结合成硫酸酯;N-甲转移酶使伯胺、肿胺及叔胺甲基化,以S-腺苷甲硫氨酸作为甲基供应体;磺胺类及芳香族氨基等在乙酰辅酶A参与下乙酰化。3、药物代谢的意义:(1)解毒,绝大多数药物通过代谢后失去药理活性,称为解毒。肝药酶活性低时,应用主要在肝灭活的药物时要特别慎重。 (2)活化,少数药物经代谢变化后效力反而增强,称为活化。4、药酶的诱导剂和抑制剂:某些药物可促进药酶对其的降解,又可促进其它药物的药酶的降解作用,长期服用可产生耐受性。有些药物能抑制药酶的活性,从而延缓药物的降解,长期应用可产生积蓄中毒。
(四)排泄:主要通过肾脏。此外还有肺、胆汁、乳汁、唾液腺、支气管腺、汗腺、肠道等。1、肾脏排泄包括肾小球滤过和肾小管排泌。肾小球滤孔约600Å,分子量<65000均可通过。肾小管排泌是主动转运过程,需要载体,肾小管上皮细胞具有两类转运系统(两种载体):有机酸转运系统,转运有机酸药物;有机碱转运系统,转运有机碱药物。有饱和现象,对同一转运系统有竞争性抑制。肾小管上皮细胞膜也具类脂结构,药物可通过脂溶扩散从肾小管重吸收回到血液中去,肾小管重吸收的主要是未离解的脂溶性药物,改变尿液pH可影响药物的离解度,能显著影响弱酸性或弱碱性药物在肾小管的重吸收;相反,增加弱酸性药物的离解度,可减少其在肾小管的重吸收,加速其排泄率。故弱酸性药物中毒时,宜用碳酸氢钠碱化尿液,加速毒物排出。肾功能不全者慎用或禁用主要经肾排泄的药物。2、从胆汁排泄的药物,除需具有一定的化学结构外,分子量要超过300才可以。分子量超过5000的大分子或蛋白质很难从胆汁排出。药物从肝细胞向胆汁的转运是主动转运过程,需有载体,有饱和现象。肝细胞至少有三个转运系统:有机酸类转运、有机碱类转运和中性化合物转运。属同一转运系统的药物,有竞争性抑制。药物由胆汁排入十二指肠后,有些从粪便排出,有些可被肠上皮细胞吸收入血液,形成“肝-肠循环”。 3、某些药物可从乳汁排泄,可能引起乳儿中毒。4、某些挥发性药物可从肺排泄。5、有些药物可从支气管排泄。6、有些可从汗腺排泄。
药物速率
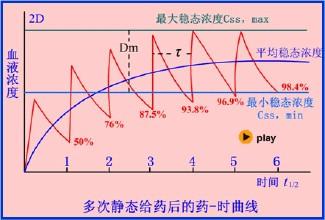
(一)、药物浓度―时间曲线:给药后药物浓度随时间迁移发生变化为纵坐标,以时间为横坐标绘制曲线图,称为药物浓度―时间曲线(见图)。由于血液是药物及其代谢物在体内吸收、分布代谢和排泄的媒介,各种体液和组织中的药物浓度与血液中的药物浓度保持一定的比例关系,而有些体液采集较困难,所以血药浓度变化最具有代表性,是最常用的样本,其次是尿液和唾液。 (二)、消除速率类型: 1、一级速率消除:单位时间内体内药物浓度按恒定比例消除。计算公式为:dC/dt ﹦―KC 2、零级速率消除:单位时间内体内药物浓度按恒定的量消除。计算公式为:dC/dt ﹦―KoC° dC/dt ﹦―Ko 3、混合速率消除:少部分药物小剂量时以一级速率转运,而在大剂量时以零级速率转运。因此描述这类药物的消除速率需要将两种速率类型结合起来,通常以米﹣曼氏方程式描述。 (三)、药动学模型:房室模型是药动学研究中广为采用的模型之一,由一个或数个房室组成,一个是中央室,其余是周边室。这种模型是一种抽象的表达方式,并非指机体中的某一个器官或组织。 (四)、药动学参数计算及意义: 1、峰浓度和达峰时间:指血管外给药后药物在血浆中的最高浓度值及其出现时间,分别代表药物吸收的程度和速度。 2、曲线下面积:指时量曲线和横坐标围成的区域,表示一段时间内药物在血浆中的相对累积量。 3、生物利用度:药物经血管外给药后能被吸收进入体循环的分量及速度。 4、生物等效性:比较同一种药物的相同或者不同剂型,在相同试验条件下,其活性成分吸收程度和速度是否接近或等同。 5、表观分布容积:指理论上药物均有分布应占有的体液容积。 6、消除速率常数:指单位时间内消除药物的分数。 7、半衰期:指血浆中药物浓度下降一半所需要的时间。 8、清除率:指单位时间内多数毫升血浆中的药物被清除。
剂量优化设计
靶浓度
合理的给药方案是使稳态血浆药物浓度(Css)达到一个有效而不产生毒性反应的治疗浓度范围,称为靶浓度(target concentration)。
维持量
在大多数情况下,临床多采用多次间歇给药或是持续静脉滴注,以使稳态血浆药物浓度维持在靶浓度。因此要计算药物维持剂量(maintenance dose)
负荷量
t1/2才能达到稳态血药浓度,增加剂量或者缩短给药间隔时间均不能提前达到稳态,只能提高药物浓度,因此如果患者急需达到稳态血药浓度以迅速控制病情时,可用负荷量(loadingdose)给药法。负荷量是指首次剂量加大,然后再给予维持剂量,使稳态血药浓度(即事先为该患者设定的靶浓度)提前产生。