芳香烃(分子中含有苯环的化合物)

VLoG
次浏览
更新时间:2023-05-22
基本信息
| 中文名 | 芳香烃 |
| 外文名 | aromatic hydrocarbon |
| 别名 | 芳烃 |
| 水溶性 | 低 |
| 用途 | 多用于制药、染料等工业 |
| 定义 | 具有芳香性的烃 |
收起
结构构成
苯的结构和表达
(1)苯的结构
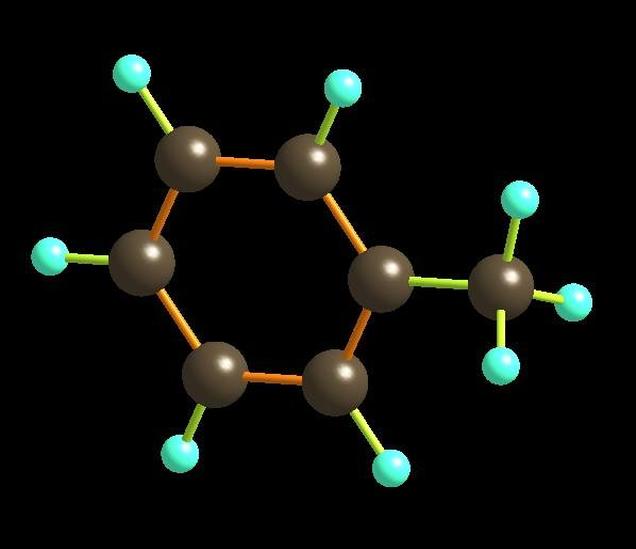
近代物理方法证明:苯分子的六个碳原子和六个氢原子都在一个平面内,因此它是一个平面分子,六个碳原子组成一个正六边形,碳键键长是均等的,约为140 pm,介于单键和双键之间。碳氢键键长为108pm,所有的键角都为120°。
(2)苯的芳香性
从结构上看,苯具有平面的环状结构,键长完全平均化,碳氢比为1。从性质上看,苯具有特殊的稳定性:环己烯的氢化热ΔH=-120kJ/mol,1,3-环己二烯的氢化热ΔH=-232kJ/mol(由于其共轭双键增加了其稳定性)。而苯的氢化热ΔH=-208kJ/mol。1,3-环己二烯失去两个氢变成苯时,不但不吸热,反而放出少量的热量。这说明:苯比相应的环己三烯类要稳定得多,从1,3-环己二烯变成苯时,分子结构已发生了根本的变化,并导致了一个稳定体系的形成。
苯还具有特殊的光谱特征。苯环上的氢处于核磁共振的低场。
上述特点说明了苯具有典型的芳香特征。
(3)苯的表达
怎样来表达苯的结构?自1825年英国物理学家和化学家Farady M(法拉第)首先从照明气中分离出苯后,人们一直在探索苯结构的表达式。科学家们提出了各种有关苯结构式的假设;其中比较有代表性的苯的结构式有:
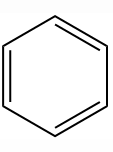
凯库勒式(凯库勒1865年提出)
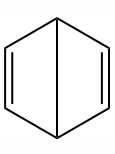
双环结构式(杜瓦1866-1867年提出)

棱形结构式(拉敦保格1869年提出)

向心结构式(阿姆斯特朗1887-1888年提出)
对位键结构式(克劳斯1888年提出)
苯的结构式
关于苯的结构及它的表达方式已经讨论了140多年了,虽然提出了各种看法,但还没有得到满意的结果,需要作进一步讨论。
联苯的结构
最简单的联苯是二联苯。在二联苯中,每个苯环都保持了苯的结构特性。连接两个苯环之间的单键可以自由旋转,但当二联苯的四个邻位氢原子都被相当大的基团取代时,单键的旋转将会受到阻碍,并产生出一对光活性异构体。
物理性质
化学性质
还原反应
Birch还原反应与苯环的催化氢化不同,它可使芳环部分还原生成环己二烯类化合物,因此Birch还原有它的独到之处,在合成上十分有用。
萘同样可以进行Birch还原。萘发生Birch还原时,可以得到1,4二氢化萘和1,4,5,8-四氢化萘。
2.催化氢化反应
苯在催化氢化( catalytic hydrogenation)反应中一步生成环己烷体系。萘在发生催化加氢反应时,使用不同的催化剂和不同的反应条件,可分别得到不同的加氢产物。蒽和菲的9、10位化学活性较高,与氢气加成反应优先在9、10位发生。
3.用金属还原
用醇和钠也可以还原萘,温度稍低时得1,4-二氢化萘,温度高时得1,2,3,4-四氢化萘。
氧化反应
1.苯及其衍生物的氧化
而且,不管侧链多长,只要和苯环相连的碳上有氢,氧化的最终结果都是侧链变成只有一个碳的羧基,如果苯环上有两个不等长的侧链,通常是长的侧链先被氧化。
2.萘、蒽和菲的氧化反应
取代反应
芳香族化合物芳核上的取代反应从机理上讲包括亲电、亲核以及自由基取代三种类型。所谓芳香亲电取代(aromatic electrophilic substitution)是指亲电试剂取代芳核上的氢。苯的亲电取代称为苯的一元素电取代,一元取代苯再在苯环上发生亲电取代称为苯的二元亲电取代。典型的芳香亲电取代有苯环的硝化、卤化、磺化、烷基化和酰基化。这些反应的反应机理大体是相似的。
芳香族化合物的硝化反应是一个十分有用的取代反应。例如:苯甲醛的硝化产物间硝基苯甲醛是生产强心急救药阿拉明的重要原料。
因为醛基易氧化,因此反应必须在低温(0℃)进行,操作时,先在浓硫酸中加入少量发烟硝酸,冷却至0℃,然后慢慢滴加苯甲醛和发烟硝酸,反应完成后,立即将产物倾倒在冰中。许多硝基化合物是炸药。广泛使用的强烈炸药TNT是2,4,6-三硝基甲苯,它是甲苯经分阶段硝化制备的,即三个硝基是在多次硝化反应中逐步引入的。
三次硝化的硝化试剂(即混合酸)浓度逐渐增高,在生产中,为节约成本,可把第三阶段硝化后的混合酸用于第二阶段硝化,第二阶段硝化后的混合酸用于第一阶段硝化。如果需要得到中间产物,反应可以在第一阶段或第二阶段中止,邻硝基甲苯和对硝基甲苯可以通过减压蒸馏或重结品分离提纯而分别获得,2,4二硝基甲苯也能通过重结晶提纯得到。
一元取代苯进行二元硝化时,已有的基团对后进入基团进入苯环的位置产生制约作用,这种制约作用即为取代基的定位效应(directing effect)。取代基的定位效应是与取代基的诱导效应、共轭效应、超共轭效应等电子效应有关的。
1.取代基的诱导效应和共轭效应
共轭效应是取代基的σ(或π)轨道上的电子云与苯环碳原子的p轨道上的电子云互相重叠,从而使σ(或π)电子发生较大范围的离域引起的,离域的结果如使取代基的σ电子向苯环迁移则发生了给电子的共轭效应,如使苯环上的π电子向取代基迁移则发生了吸电子的共轭效应。产生给电子共轭效应的取代基有:
—NR2>—OR>—F,—O->—OR,—F>—Cl>—Br>—I
绝大多数取代基既可与苯环发生诱导效应,也可发生共轭效应,最终的表现是两者综合的结果。大部分取代基的诱导效应与共轭效应方向是一致的,但有的原子或基团的诱导效应与共轭效应方向不一致。例如,卤素的电负性比较大,它具有吸电子诱导效应,卤苯的卤原子的p轨道与苯环碳上的p轨道平行重叠,卤原子的孤电子对离域到苯环上,发生给电子的共轭效应,但总的结果是吸电子的诱导效应大于给电子的共轭效应,因此卤素是吸电子基,它使苯环的电子云密度降低。取代基的综合电子效应可以从取代苯的偶极矩大小和方向上表现出来。
在烷基苯中,烷基与苯环不发生共轭作用,但烷基的C-H中σ电子与苯的π电子能发生σ-π超共轭作用,烷基的超共轭作用有微弱的给电子能力。
硝基苯硝化的反应式及实验数据如下所示:
将上面的式子与苯的硝化对比,可以得出下述结论:
(1)硝基苯比苯难硝化得多,需要用比较强的条件,例如提高反应温度、增加酸的浓度等来实现。
(2)硝基苯硝化时,主要得到间位产物,邻、对位产物极少。
硝基苯比苯难硝化的原因是:苯环的硝化是一个亲电取代反应,硝化反应的机理表明:整个反应的关键一步是硝基正离子进攻苯环形成中间体碳正离子。在硝基苯中,因氧、氮的电负性均大于碳,因此硝基有吸电子的诱导效应,叉因为硝基的π轨道与苯环的离域π轨道形成一个π-π共轭体系,使苯环的π电子云也向硝基迁移,所以硝基是一个具有强吸电子诱导效应和吸电子共轭效应的取代基。它使苯环的电子云密度有较大程度的下降,这一方面增加了硝基正离子进攻苯环的难度,同时也降低了反应过程中产生的中间体碳正离子的稳定性,所以硝基苯比苯难硝化。
3.甲苯的硝化反应
甲苯硝化的反应式及实验数据如下所示:
甲苯完全硝化,可直接得到三硝基甲苯(TNT)。

实验结果表明:①甲苯比苯容易硝化;②甲苯硝化时,主要得到邻位和对位产物。
甲苯比苯容易硝化的原因是:甲基具有微弱的给电子超共轭效应,这种超共轭效应使苯环上的电子云密度有所增加,这一方面使硝基正离子更容易进攻苯环,同时也使反应过程中产生的中间体碳正离子的电荷得到分散而稳定。所以甲苯比苯更易硝化。但甲基的给电子能力是很弱的,因此它对苯环的活泼性影响较弱。
氯苯硝化的反应式及实验数据如下所示:
实验结果表明:①氯苯比苯难以硝化;②氯苯硝化时主要得到邻、对位取代产物。
氯苯比苯难以硝化的原因是:氯原子的吸电子诱导效应比给电子共轭效应大,总的结果使苯环上的电子云密度降低,这一方面使硝基正离子不易进攻苯环,另一方面使反应过程中产生的中间体碳正离子更不稳定,反应时过渡态势能增大,所以氯苯比苯难硝化。
铁粉与氯气或溴反应可生成三氯化铁或三溴化铁,因此也可以用铁粉代替三氯化铁、三溴化铁做催化剂。反应时,首先是卤素与苯形成π络合物,光谱和X射线衍射法都已证明了π络合物的存在。在形成π络合物时,氯分子的键没有异裂,然后在缺电子的Lewis酸的作用下,氯分子键极化,进而发生键的异裂,生成活性中间体碳正离子,然后失去氢生成氯苯。
苯的溴化也可直接进行,但速率很慢。
卤素由于活泼性不同,发生卤化反应时,反应性也不同。最大的差别是氟太活泼,不宜与苯直接反应,因直接反应时,只生成非芳香性的氟化物与焦油的混合物。大量的苯在四氯化碳溶液中,与含有催化量氟化氢的二氟化氙反应,可制得产率为68%的氟苯。
如果是较长的侧链,卤化反应也可以在别的位置发生,但是σ位的选择性最高,这是因为苯甲型自由基最稳定的缘故。
傅—克反应
Friedel(傅瑞德尔)- Crafts(克拉夫兹)反应,简称傅一克反应。有机化合物分子中的氢被烷基(-R)取代的反应称为烷基化反应,被酰基取代的反应称为酰基化反应。苯环上的烷基化反应和酰基化反应统称为傅克反应。
1.傅—克烷基化反应
傅一克烷基化反应(Friedel-Crafts alkylation)的反应机理与磺化、硝化类似,首先在催化剂的作用下产生烷基碳正离子,它作为亲电试剂向苯环进攻,形成碳正离子,然后失去一个质子生成烷基苯。
卤代烷、烯烃、醇、环氧乙烷等在适当催化剂的作用下都能产生烷基碳正离子,卤代烷、烯烃、醇是常用的烷基化试剂。最初用的催化剂是三氯化铝,后经证明,许多Lewis酸同样可以起催化作用。
2.傅一克酰基化反应
常用的催化剂是三氯化铝。由于AlCl3能与羰基络合,因此酰化反应的催化剂用量比烷基化反应多,含一个羰基的酰卤为酰化试剂时,催化剂用量要多于1 mol反应时,酰卤先与催化剂生成络合物,少许过量的催化剂再发生催化作用使反应进行。如用含两个羰基的酸酐为酰化试剂,因同样原因,催化剂用量要多于2 mol。
氯甲基化反应与Gattermann—Koch反应
1.氯甲基化反应
氯化苄(henzyl chloride)也称为苄氯,可通过苯与甲醛、氯化氢在无水氯化锌作用下反应制得,此反应称为氯甲基化( chloromethylation)反应。苄氯上的氯十分活泼,可以转化为各种有用的化合物。
2.Gattermann—Koch反应
在Lewis酸及加压情况下,芳香化合物与等物质的量的一氧化碳和氯化氢的混合气体发生作用可以生成相应的芳香醛。在实验室中则用加入氯化亚铜来代替工业生产的加压方法。因氯化亚铜可与一氧化碳络合,使之活性增高而易于发生反应。
电取代经验规律
苯的多元亲电取代是指二元取代苯或含有更多取代基的苯衍生物进行亲电取代反应,其中最简单的是二元取代苯的进一步取代。和苯的二元取代一样,苯环上已有的取代基对新进入苯环的取代基也有定位作用。二元或多元取代苯的定位问题比一元取代苯复杂。总的来说,最终反映出来的定位作用实际上是苯环上已有取代基的综合作用,若已有取代基的定位作用一致,则它们的作用可以互相加强。
两个取代基中间的位置一般不易进入新基团。
当已有取代基的定位作用不一致时,可参照下列经验规则:
(1)多数情况下,活化基团的作用超过钝化基团的作用。
(2)强活化基团的影响比弱活化基团的影响大。
(3)两个基团的定位能力没有太大差别时,主要得到混合物。
巧妙地利用取代基的定位效应,合理地确定取代基进入苯环的先后次序可以有效地合成芳香族化合物。例如,由苯合成邻硝基氯苯要先氯化后硝化,而合成间硝基氧苯则要先硝化而后氯化。又如,用甲苯制备3-硝基-5-溴苯甲酸时,因为三个取代基互为间位,因此要优先引入间位定位基,即要先氧化,再硝化,最后溴化。而用甲苯制备2,4一二硝基苯甲酸,则要先硝化再氧化。
除取代基的定位效应外,反应温度、溶剂、催化剂、新进入取代基的极性、体积等众多因素对取代基进入苯环的位置也都有影响。例如,甲苯在不同温度下进行磺化,所得产物中各异构体的产率如下所示:
反应温度/℃ | 邻/% | 对/% | 间/% |
100 | 13 | 79 | 8 |
0 | 50 | 43 | 4 |
又如溴苯分别用三氯化铝和三氯化铁做催化剂进行溴化,所得异构体的产率分别为:
催化剂 | 邻/% | 对/% | 间/% |
AlCl3 | 8 | 62 | 30 |
FeCl3 | 13 | 85 | 2 |
再如溴苯氯化,产物中邻、对、间位异构体分别为:42%,51%.7%;随着进入基团体积的增大,邻位异构体产量减少,对位异构体增多,这主要是空间效应的结果。因此在进行反应和合成时,要全面考虑问题。
亲电取代反应
在正常情况下,萘比苯更易发生典型的芳香亲电取代反应,硝化和卤化反应主要发生在α位上。
为什么取代反应主要发生在α位上?共振理论认为:取代基进攻α位形成的碳正离子中间体有两个稳定的含有完整苯环结构的极限式,而进攻卢位形成的碳正离子中间体只有一个稳定的含有完整苯环结构的极限式,所以前者比后者稳定。显然,稳定碳正离子相对应的过渡态势能也相对较低,所以进攻α位,反应活化能较小,反应速率快。
上述现象表明,与萘的硝化、卤化反应一样,生成α-萘磺酸比生成β-萘磺酸活化能低,低温条件下提供能量较少,所以主要生成α-萘磺酸。但磺化反应是可逆的,由于,α-磺基与异环的α-H处于平行位置,空阻较大,不稳定,随着反应温度升高,α-萘磺酸的增多,α-磺化反应的逆向速率将逐渐增加;另外,温度升高也有利于提供β-磺化反应所需的活化能,使其反应速率也加大,β-磺基与邻近的氢距离较大,稳定性好,其逆向反应速率很慢,所以α-萘磺酸逐渐转变成β-萘磺酸。
萘的酰化反应既可以在α位发生,也可以在β位发生,反应产物与温度和溶剂很有关系。
如果第一取代基(G)在β位时,有时6位也能发生取代反应,因为6位也可以被认为是G的对位。
间位取代基使环钝化,因此取代反应主要发生在异环的α位。
但是,磺化和傅一克反应常在6,7位发生,生成热力学稳定产物。
蒽比苯、萘更易发生亲电取代反应,除磺化反应在1位发生外,硝化、卤化、酰化时均得9-取代蒽,取代产物中常伴随有加成产物。
菲的9,10的化学活性很高,取代首先在9,10位发生。
此外菲的1,2,3,4,10和5,6,7,8,9是对应的,所以应有五种一元取代产物。
制备方法
来源
煤和石油是制备一些简单芳香烃如苯、甲苯等的原料。而这些简单芳香烃又是制备其它高级芳香族化合物的基本原料。当煤在无氧条件下加热至1000℃,煤分子通过热分馏产生煤焦油。而从煤焦油中可以产生苯、甲苯、二甲苯、萘和其它芳香化合物。与煤不同,石油中含有大量的烷烃和少量芳香化合物。石油馏分中主要含有环烷烃和链烃,将它们转化为芳香烃的主要方法是重整和芳构化。芳构化是指含六元环的脂环族化合物在铂、钯、镍等催化剂存在下,加热脱氢,生成芳香族化合物的过程。重整包括链烃裂解、异构化、关环、扩环、氢转移、烯烃吸氢等过程。重整一般都是在催化剂作用下进行的,常用的催化剂有铂、铜等。石油工业化学中一个重要的反应称临氢重整(或称铂重整),就是在氢存在下,以铂为催化剂使分子结构重新安排,例如在Pt,SiO2/Al2O3,500℃,1~4 MPa及H2存在的反应条件下进行反应。临氢重整反应过程很复杂,因为反应体系是混合物。
傅一克反应作用
合成过程中要使用两次傅一克酰基化反应,第一次生成γ-氧代γ苯基丁酸,经Cemmensen还原后,再进行第二次酰化关环,然后再经一次Clemmensen还原及硒脱氢后得萘。
蒽醌是合成一大类蒽醌染料的重要中间体,它的工业合成方法是经过两次傅一克酰基化反应得到蒽醌。蒽醌再经还原脱氢后,得到蒽。
γ-芳基丁酸在多磷酸(或85% H2S04)作用下,加热环化成六元环酮,环酮用锌汞齐、盐酸还原后,再用硒加热脱氢得到多环芳香族化合物。这个合成多环化合物的重要方法称为Haworth(哈沃斯)反应。用苯合成萘、由萘合成菲的反应实例说明,Haworth反应在合成稠环化合物中十分有用。
蔻合成方法
蔻(coronene)在自然界中不存在,可用多种方法合成。其中一个方法是利用傅一克反应及硒脱氢反应而实现的。用2,7二甲萘通过N一溴代丁二酰亚胺进行苯甲型的溴化,两个甲基的氢各被一个溴取代,然后用Wurtz反应将两分子缩合,即得到一个十四元的环状化合物。在三氯化铝的作用下即行关环。这步反应的过程和芳香烃被烯烃烷基化类似。最后用硒脱氢即得到蔻。
主要危害
苯是一种应用广泛的有机溶剂,是黏合剂、油性涂料、油墨等的溶剂。短时间内吸入大量苯蒸气可引起急性中毒。急性苯中毒主要表现为中枢神经系统麻醉,甚至导致呼吸心跳停止。长期反复接触低浓度的苯可引起慢性中毒,主要是对神经系统、造血系统的损害,表现为头痛、头晕、失眠,白血球持续减少、血小板减少而出现出血倾向,甚至诱发白血病。我国规定操作车间内空气中苯的浓度不得超过40mg·m,居室内空气中苯含量平均每小时不得超过0.09mg·m.制鞋、皮革加工业、箱包、家具制造中使用的黏胶剂,喷漆、油漆工作中使用的溶剂都含有苯或苯的同系物,因此从事上述职业的人群要加强防范,避免苯中毒。虽然环芳烃对人体有害,但是人类却对一种原始的烹饪方式情有独钟,那就是烧烤(这可能是由于祖先遗传给我们的行为因子造成的,在有些时候竟然使我们对环芳烃产生一种愉悦感)。