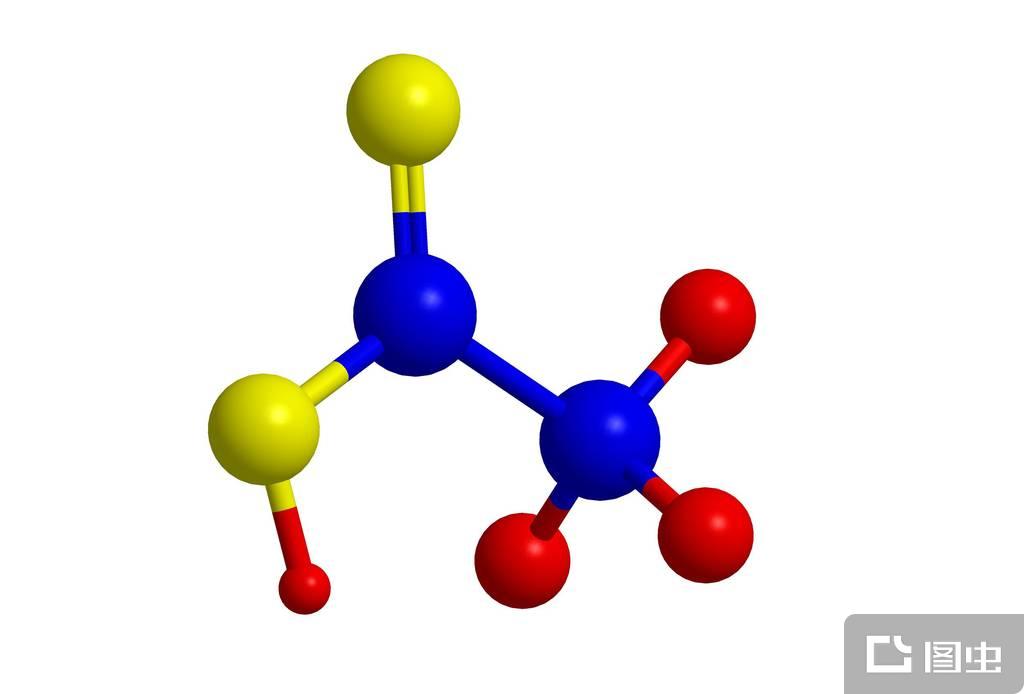
配位化合物(具有特征化学结构的化合物)

VLoG
次浏览
更新时间:2023-05-23
配位化合物
具有特征化学结构的化合物

基本信息
| 中文名 | 配位化合物 |
| 外文名 | Coordination complex/metal complex |
| 别名 | 错合物、络合物 |
| 简称 | 配合物 |
高中教材一般定义
术语
讨论经典配位化合物时,常会提到以下的术语:
配位键、配位共价键:配位化合物中存在的化学键,由一个原子提供成键的两个电子,成为电子给予体,另一个成键原子则成为电子接受体。参见酸碱反应和路易斯酸碱理论。配位单元:化合物含有配位键的一部分,可以是分子或离子。配离子:含有配位键的离子,可以是阳离子或阴离子。内界、外界:内界指配位单元,外界与内界相对。配体、配位体、配位基:提供电子对的分子或离子。配位原子:配体中,提供电子对的原子。中心原子、金属原子:一般指接受电子对的原子。配位数:中心原子周围的配位原子个数。螯合物:含有螯合配体的配合物。
此外,含有多个中心原子的配合物称为多核配合物,连接两个中心原子的配体称为桥联配体,以羟基桥联的称为羟联,以氧基桥联的称为氧联。
配位化合物
在配合物中,中心原子与配位体之间共享两个电子,组成的化学键称为配位键,这两个电子不是由两个原子各提供一个,而是来自配位体原子本身,例如[Cu(NH3)4]SO4中,Cu与NH3共享两个电子组成配位键,这两个电子都是由N原子提供的。形成配位键的条件是中心原子必须具有空轨道,而过渡金属原子最符合这一条件。
历史
人们很早就开始接触配位化合物,当时大多用作日常生活用途,原料也基本上是由天然取得的,比如杀菌剂胆矾和用作染料的普鲁士蓝。最早对配合物的研究开始于1798年。法国化学家塔萨厄尔首次用二价钴盐、氯化铵与氨水制备出CoCl3·6NH3,并发现铬、镍、铜、铂等金属以及Cl、H2O、CN、CO和C2H4也都可以生成类似的化合物。当时并无法解释这些化合物的成键及性质,所进行的大部分实验也只局限于配合物颜色差异的观察、水溶液可被银离子沉淀的摩尔数以及电导的测定。对于这些配合物中的成键情况,当时比较盛行的说法借用了有机化学的思想,认为这类分子为链状,只有末端的卤离子可以离解出来,而被银离子沉淀。然而这种说法很牵强,不能说明的事实很多。
1893年,瑞士化学家维尔纳总结了前人的理论,首次提出了现代的配位键、配位数和配位化合物结构等一系列基本概念,成功解释了很多配合物的电导性质、异构现象及磁性。自此,配位化学才有了本质上的发展。维尔纳也被称为“配位化学之父”,并因此获得了1913年的诺贝尔化学奖。
基本组成

配位化合物
[Cu(NH3)4]SO4。中心原子可以是带电的离子,如[Cu(NH3)4]SO4中的Cu。配体给出孤对电子或多个不定域电子,中心原子接受孤对电子或多个不定域电子,组成使二者结合的配位键。例如,K4[Fe(CN)6]、[Cu(NH3)4]SO4、[Pt(NH3)2Cl2]和[Ni(CO)4]都是配合物。其中:CN:-、∶NH3、和∶CO∶是配体,皆有孤对电子(∶),Fe、Cu、Pt和Ni是中心原子,皆可接受孤对电子。配体和中心原子组成配位本体,列入方括弧中。配合物在溶液中发生部分离解,但仍趋向保持其本体。周期表中所有金属均可作为中心原子,其中过渡金属(见过渡元素)比较容易形成配合物。非金属也可作为中心原子。配体分为单齿配体和多齿配体两种。单齿只有一个配位原子,例如CN、CO、NH3和Cl均是单齿配体,配位原子分别是C、N和Cl,它们直接与中心原子键合。多齿有两个或两个以上配位原子:乙二胺H2NCH2CH2NH2是双齿配体,配位原子是两个N原子;乙二胺四乙酸根(简称EDTA4-)(-OOCCH2)2N-CH2-CH2-N(CH2COO-)2是六齿配体,配位原子是两个N和四个羧基上的O。配体为负离子或中性分子,偶尔也有正离子(如NH2NH)。带电荷的配位本体称为配离子,带正电荷的配离子称配阳离子,带负电荷的称配阴离子。配离子的电荷为金属离子和配体所带电荷之和,例如Fe和6CN配位产生[Fe(CN)6]配阴离子,Cu和4NH3产生[Cu(NH3)4]配阳离子,它们各与带相反电荷的阳离子或阴离子组成配合物。中性配位本体就是配合物,例如Pt和2NH3及2Cl产生[Pt(NH3)2Cl2];Ni和4CO产生[Ni(CO)4]。配合物可为单核或多核,单核只有一个中心原子;多核有两个或两个以上中心原子。上述配合物均为单核配合物;多核配合物如[(CO)3Fe(CO)3Fe(CO)3]。
命名方法
①命名配离子时,配位体的名称放在前,中心原子名称放在后。②配位体和中心原子的名称之间用“合”字相连。③中心原子为离子者,在金属离子的名称之后附加带圆括号的罗马数字,以表示离子的价态。④配位数用中文数字在配位体名称之前。⑤如果配合物中有多种配位体,则它们的排列次序为:阴离子配位体在前,中性分子配位体在后;无机配位体在前,有机配位体在后。不同配位体的名称之间还要用中圆点分开。根据以上规则,〔Cu(NH3)4〕SO4称硫酸四氨合铜(Ⅱ),〔Pt(NH3)2Cl2〕称二氯·二氨合铂(Ⅱ),K〔Pt(C2H4)Cl3〕称三氯·(乙烯)合铂(Ⅱ)酸钾。实际上,配合物还常用俗名,如K4〔Fe(CN)6〕称黄血盐,K3〔Fe(CN)6〕称赤血盐,Fe4〔Fe(CN)6〕3称普鲁士蓝。
在命名配位化合物时,一般遵循中文IUPAC命名法,命名规律有:
- 桥联配体前要加注μ;η则表示配体有n个原子与中心原子键结(n即为配体的哈普托数)。对于可能产生键合异构的配合物,需在配体后注明配位原子。
配合物 | 命名 |
[NiCl4]- | 四氯合镍酸(II)根离子 |
[Cu(NH3)Cl5]3- | 五氯·一氨合铜酸(II)根离子 |
二氰·二(乙二胺)合镉(II) | |
[Co(NH3)5Cl]SO4 | 硫酸一氯·五氨合钴(III) |
Fe2Cl6(氯化铁二聚体) | 四氯二-μ-氯合二铁(III) |
展开表格
命名规则
一、配合物的命名(供高中生学习使用)
(1)配合物的命名,关键在于配合物内界(即配离子)的命名
中心离子的化合价由外界离子电荷/配位体电荷按配合物电荷为零计算得到,在中心离子后面用小括号加罗马数字表示。
例子略。
(2)配合物可看作盐类,若内界是阳离子,外界必是阴离子;若内界是阴离子,外界必是阳离子。可按盐的命名方法命名,自右向左可命名为某酸某或某化某。
如果配合物中有多种配位体,则它们的排列次序为:阴离子配位体在前,中性分子配位体在后;无机配位体在前,有机配位体在后。
例子略。
配合物溶于水易电离为内界配离子和外界离子,而内界的配位体和中心原子通常不能电离。
Eg:[Co(NH3)5Cl]Cl2——〉[Co(NH3)5Cl]+2Cl,有三分之一的Cl不能被电离。
二、详细说明(供学术使用)
(1)在配合物中
先阴离子,后阳离子,阴阳离子之间加“化”字或“酸”字,配阴离子看作是酸根。
(2)在配位单元中
①先配体后中心离子(或原子),配体与中心离子(或原子)之间加“合”字。
②配体前面用一、二、三等表示该配体个数。“一”可省略。如易引起误解则需给配体加上括号。
③几种不同的配体之间用“˙”隔开。
④中心离子后面加“()”,用罗马数字表示中心离子(或原子)的价态。
(3)配体的先后顺序
下述的每条规定均在其前一条的基础上
①先无机配体后有机配体
②先阴离子类配体,后阳离子类配体,最后分子类配体
如K[PtCl3(NH3)]三氯˙一氨合铂(Ⅱ)酸钾
③同类配体中,按配位原子的元素符号在英文字母表中的次序分出先后
如[Co(NH3)5H2O]Cl3三氯化五氨˙一水合钴(Ⅲ)
④配位原子相同,配体中原子个数少的在前
⑤配体中原子个数相同,则按和配位原子直接相连的配体中的其他原子的元素符号在英文字母表中的次序。如NH和NO,则NH在前。
基本分类
按配位体分类,可有:
①水合配合物。为金属离子与水分子形成的配合物,几乎所有金属离子在水溶液中都可形成水合配合物,如〔Cu(H2O)4〕、〔Cr(H2O)6〕。
③氨合配合物。金属离子与氨分子形成的配合物,如〔Cu(NH3)4〕SO4。
④氰合配合物。金属离子与氰离子形成的配合物,如K4〔Fe(CN)6〕。
配位化合物
⑤金属羰基化合物。金属与羰基(CO)形成的配合物。如〔Ni(CO)4〕。
按中心原子分类,可有:
①单核配合物。只有一个中心原子,如K2〔CoCl4〕。
②多核配合物。中心原子数大于1,如〔(H3N)4Co(OH)(NH2)Co(H2NCH2CH2NH2)2〕Cl4。
按成键类型分类,可有:
①经典配合物。金属与有机基团之间形成σ配位键,如〔Al2(CH3)6〕。
按学科类型分类,可有:
①无机配合物。中心原子和配位体都是无机物。
②有机金属配合物。金属与有机物配位体之间形成的配合物。
- 传统配位化合物由一个以上的配离子(也叫离子复合物)形成,配位键中的电子“几乎”全部由配体提供。典型的配体包括H2O、NH3、Cl、CN和en。
- 例子:[Co(EDTA)]、[Co(NH3)6]Cl3、[Fe(C2O4)3]K3和[Cr(H2O)6]Cl3。
与配位化学有交盖的化学分支如:
- 原子簇化学——用金属原子作配体,如Ru3(CO)12。
主要性质
稳定性
通常,配位化合物的稳定性主要指热稳定性和配合物在溶液中是否容易电离出其组分(中心原子和配位体)。配位本体在溶液中可以微弱地离解出极少量的中心原子(离子)和配位体,例如〔Cu(NH3)4〕可以离解出少量的Cu和NH3:
K越大,配合物越稳定,即在水溶液中离解程度小。
配合物在溶液中的稳定性与中心原子的半径、电荷及其在周期表中的位置有关,也就是该配合物的离子势:φ=Z/rφ为离子势Z为电荷数r为半径。过渡金属的核电荷高,半径小,有空的d轨道和自由的d电子,它们容易接受配位体的电子对,又容易将d电子反馈给配位体。因此,它们都能形成稳定的配合物。碱金属和碱土金属恰好与过渡金属相反,它们的极化性低,具有惰性气体结构,形成配合物的能力较差,它们的配合物的稳定性也差。
配合物的稳定性符合软硬亲和理论,即软亲软、硬亲硬。
基本结构
构型
配位化合物的构型由配位数所决定,也就是化合物中心原子周围的配位原子个数。配位数与金属离子和配体的半径、电荷数和电子构型有关,一般在2-9之间,镧系元素和锕系元素的配合物中常会出现10以上的配位数。
把围绕中心原子的配位原子看作点,以线连接各点,就得到配位多面体。配位数与配合物构型的关系列在下表:
展开表格
五配位中,常常涉及到三角双锥和四方锥两种构型的互变,因此,很大一部分五配位化合物的结构是介于这两个结构之间的一种中间结构。六配位的化合物除极其常见的八面体外,也有可能是三角棱柱结构,例如单核配合物[Re(S2C2Ph2)3]即属于这一类。七配位中,配合物还可能是单帽八面体或单帽三角棱柱体结构。
更高配位数的化合物中,八配位的可以是四方反棱柱体、十二面体、立方体、双帽三角棱柱体或六角双锥结构;九配位的可以是三帽三角棱柱体或单帽四方反棱柱体结构;十配位的可以是双帽四方反棱柱体或双帽十二面体结构;十一配位的化合物很少,可能是单帽五角棱柱体或单帽五角反棱柱体;十二配位的如[Ce(NO3)6],为理想的二十面体;十四配位的为双帽六角反棱柱体。再高的配位数非常罕见,如PbHe15,该离子中铅的配位数至少为15。
异构现象
立体异构
几何异构
从空间关系上考虑,顺式(cis-)是指相同的配体处于邻位,反式(trans-)是指相同的配体处于对位。八面体[MA3B3]的两种异构体中,面式(fac-)或顺-顺式指3个A和3个B各占八面体的三角面的顶点,经式(mer-)或顺-反式是指3个A和3个B在八面体外接球的子午线上并列。
trans-[CoCl2(NH3)4]
fac-[CoCl3(NH3)3]
mer-[CoCl3(NH3)3]
不对称双齿配体的平面正方形配合物[M(AB)2]也有可能有几何异构现象,结构类似于上面的顺铂。
多核配合物也有几何异构现象。例如,Pt(II)的双核配合物[Pt2(PPr3)2(SEt)2Cl2]的顺反异构体都已制得,且室温下其苯溶液都是稳定的。但反式在热的或冷的苯溶液中加入痕量三丙基膦作催化剂就能完全转变为顺式。
光学异构
光学异构是立体异构的另一种形式,两种光学异构体会使平面偏振光发生等量但不同方向的偏转,因此又称旋光异构或对映异构。大多数配合物在溶液中都会逐渐失去旋光性,这一过程称为消旋作用。根据具体情况的不同,消旋机理可能是分子间或分子内的。
最简单的配合物光学异构体为四面体型,中心原子与四个不同的基团相连,分子不能与镜像重合。例如[Be(C6H5COCHCOCH3)2]。而对于八面体构型的配合物而言,光学异构主要发生在以下几种情况下:
- [M(AA)2X2]型,如[Rh(en)2Cl2]。
- [M(AB)3]型,如[Co(gly)3]。
- [M(AA)B2X2]型,如[Co(en)(NH3)2Cl2]。
Λ-[Fe(ox)3]
Δ-[Fe(ox)3]
Λ-cis-[CoCl2(en)2]
Δ-cis-[CoCl2(en)2]
结构异构
结构异构是化学式相同,但原子排列次序不同的异构体,主要可分为以下几类:
[Co(NH3)5(NO2)]的两种键合异构体。
- 键合异构:配体通过不同的配位原子与中心原子配位。配体称作两可配体,此类配体含有两个以上含孤对电子的原子,可分别与中心原子配位。常见的两可配体有:NO2、SCN和CN。
- 配位体异构:互为同分异构体的配体所形成的类似配合物,如1,3-二氨基丙烷与1,2-二氨基丙烷分别形成的钴配合物[Co(H2N-CH2-CH2-CH2-NH2)Cl2]、[Co(H2N-CH2-CH(-NH2)-CH3)Cl2]。
- 离子异构:配合物有相同分子式但不同的配位阴离子,因此水溶液中产生的离子不同,如[Co(NH3)5SO4]Br和[Co(NH3)5Br]SO4。
- 溶剂合异构:配合物中水所处的位置不同,有内界与外界的差异,例如[Co(H2O)6]Cl3和[Cr(H2O)5Cl]Cl·H2O。
- 配位异构:阳离子和阴离子都是配离子,且配体可以互相交换成分。例子有:[Co(NH3)6][Cr(CN)6]和[Cr(NH3)6][Co(CN)6]、[Cr(NH3)6][Cr(SCN)6]和[Cr(SCN)2(NH3)4][Cr(SCN)4(NH3)2],以及[Pt(NH3)4][PtCl6]和Pt(NH3)4Cl2][PtCl4]。
- 聚合异构:是配位异构的一种,用以表示配合物相对分子质量上的倍数关系,与聚合反应中的“聚合”并不类同。例如,[Co(NH3)6][Co(NO2)6]可看作[Co(NH3)3(NO2)3]的二聚体。
理论
配位化合物的化学键理论,主要研究中心原子与配体之间结合力的本性,用以说明配合物的物理及化学性质,如磁性、稳定性、反应性、配位数与几何构型等。配合物的理论起始于静电理论。而后西季威克与鲍林提出配位共价模型,也就是应用配合物中的价键理论,统治了这一领域二十余年,可以较好地解释配位数、几何构型、磁性等一些性质,但对配合物的颜色和光谱却无能为力。
价键理论认为,配体提供的孤对电子进入了中心离子的空原子轨道,使得配体与中心离子共享这两个电子。配位键的形成经历了三个过程:(激发)、杂化和成键,其中杂化也称轨道杂化,是能量相近的原子轨道线性组合成为等数量且能量简并杂化轨道的过程。由此还可衍生出外轨/内轨型配合物的概念,从而通过判断配合物的电子构型及杂化类型,就可以得出配合物的磁性、氧化还原反应性质以及几何构型。对于很多经典配合物来说,价键理论得出的结果还是比较贴近事实的。
晶体场理论将配体看作点电荷,并将配位键当作离子键处理,可看作是静电理论的延伸。并且,它以不同几何构型中,配体对不同空间取向的d轨道的作用作为切入点,得出不同取向d轨道会发生能级分裂,并建立起分裂能及晶体场稳定化能的概念,以推测配合物的电子组态及稳定性。晶体场理论可以很好地解释配合物的颜色、热力学性质和配合物畸变等现象,但不能合理解释配体的光谱化学序列,也不能很好地应用于特殊高/低价配合物、夹心配合物、羰基配合物和烯烃配合物。
配位场理论结合了分子轨道理论与晶体场理论。它在理论上更加严谨,然而定量计算则很困难,计算过程中不得不引进近似处理,因而也只能得到近似的结果。
反应
式中X为被取代的配体,通常称做离去基团;Y为取代基团,通常称为进入基团。这类配体交换反应的速率相差很大,有些反应在10秒内就可完成,而有的反应则需要数月。对于活性的差异有一个人为的规定,认为在浓度约为0.1M,温度25°C时,半衰期大于一分钟的配合物属于所谓“惰性”配合物,反之则称为活性配合物。
价键理论和配位场理论都对这类反应的速率差异做了解释,一般存在以下的规律:
- 中心金属离子电荷的增加,会使反应速率降低;
- 中心离子为d、d、d、d、d构型,高自旋d、d、d构型和高自旋d构型的配合物,对于配体交换反应都是活性的;
- 中心离子为d、d构型,或低自旋的d、d、d构型时,对于配体交换反应是惰性的。
此外,反应速率还与溶剂、配体的种类和排列有很大关系。
配位反应可视同路易斯酸碱理论中的酸碱反应:金属离子为酸提供空轨道,配体提供电子对为碱,过渡金属与配体的反应常伴随着颜色的变化。例如,将HCl加入[Cu(NH3)4]依序产生[Cu(H2O)4](淡蓝色)、[CuCl(H2O)3]、[CuCl2(H2O)2]、[CuCl3(H2O)]、[CuCl4]、[Cu(NH3)4](深蓝色);再如,将过量的氨水加入[Cu(H2O)4]颜色立即由淡蓝色转变成深蓝色:
氧化还原反应
配位化合物的氧化还原反应包含两种类型,一种是中心原子与配体之间的氧化还原反应,另一种则是两个配合物之间的氧化还原反应。后者又可分为两类:
- 桥式机理、内层反应机理:两个金属原子同时连接在一个桥联配体上,组成过渡态。
反应是以外层机理进行,还是以内层机理进行,与配合物的结构有关。对配体交换反应呈惰性、没有桥联配体或电子转移活化能很低的配合物,它们的机理以外层机理为主。对配体交换反应活性的配合物主要发生桥式机理,桥式机理所需克服的能垒较外层反应机理低很多,因为桥联配体传递电子降低了电子穿透配体外层和水化层的能量。
氧化还原反应中还有两类反应较特殊:
- 双电子转移反应:反应中氧化态的改变为±2,机理为桥式机理的可能性较大。
- 非互补反应:氧化剂与还原剂的价态改变不相等,一般反应机理分几步进行。
亨利·陶布为配合物的氧化还原反应研究做了很多贡献,也因此获得了1983年的诺贝尔化学奖。
应用
配位化合物的应用包括:
分析化学中,配合物可用于:
- 离子的分离:通过生成配合物来改变物质的溶解度,从而与其它离子分离。例如以氨水与AgCl、Hg2Cl2和PbCl2反应来分离第一族阳离子:
- 金属离子的滴定:例如,定量测定溶液中Fe的含量时,指示剂为深红色的[Fe(phen)3]。
- 掩蔽干扰离子:用生成配合物来消除分析实验中会对结果造成干扰的因素。比色法测定Co时会受到Fe的干扰,可加入F与Fe生成无色的稳定配离子[FeF6],以掩蔽Fe:
工业生产中:
- 制镜:以银氨溶液为原料,利用银镜反应,在玻璃后面镀上一层光亮的银涂层。
- 硬水软化
参考资料
[1]
配位化合物概述 · 配位化合物概述[引用日期2021-07-29]